Here’s what McElreath says about multicollinearity
Some fields actually teach students to inspect pairwise correlations before fitting a model, to identify and drop highly correlated predictors. This is a mistake. Pairwise correlations are not the problem. It is the conditional associations—not correlations—that matter. (emphasis added)
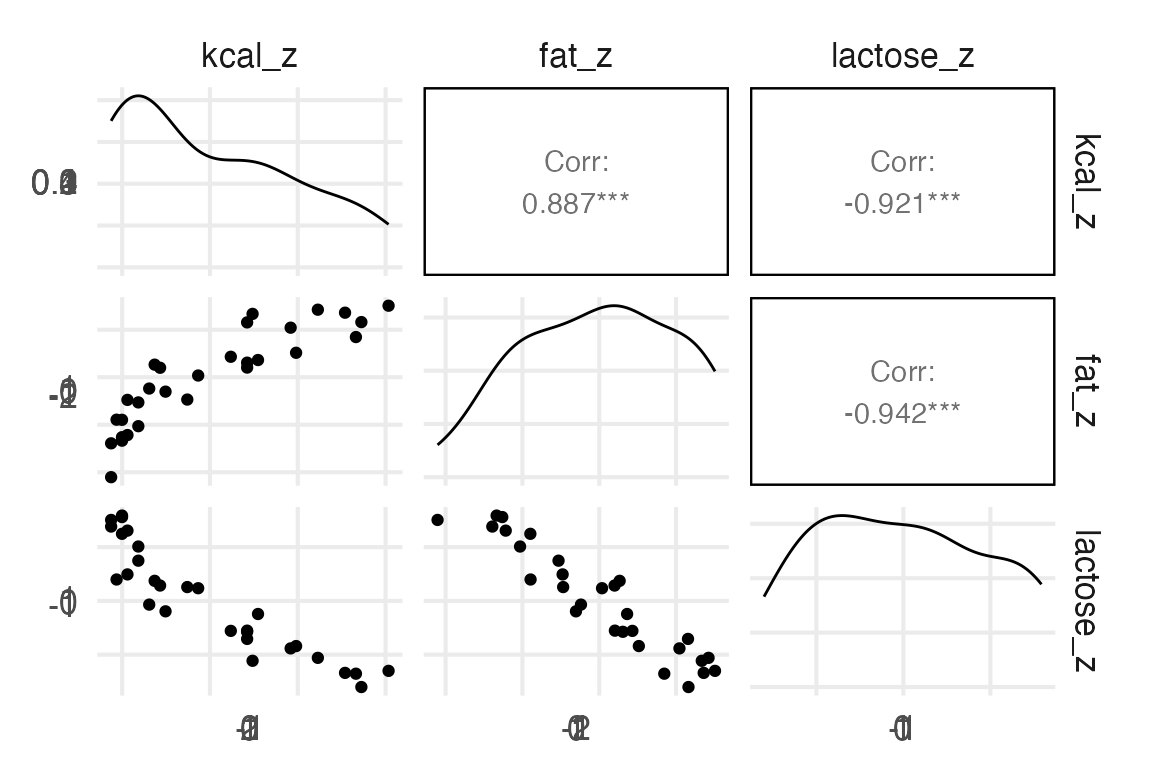
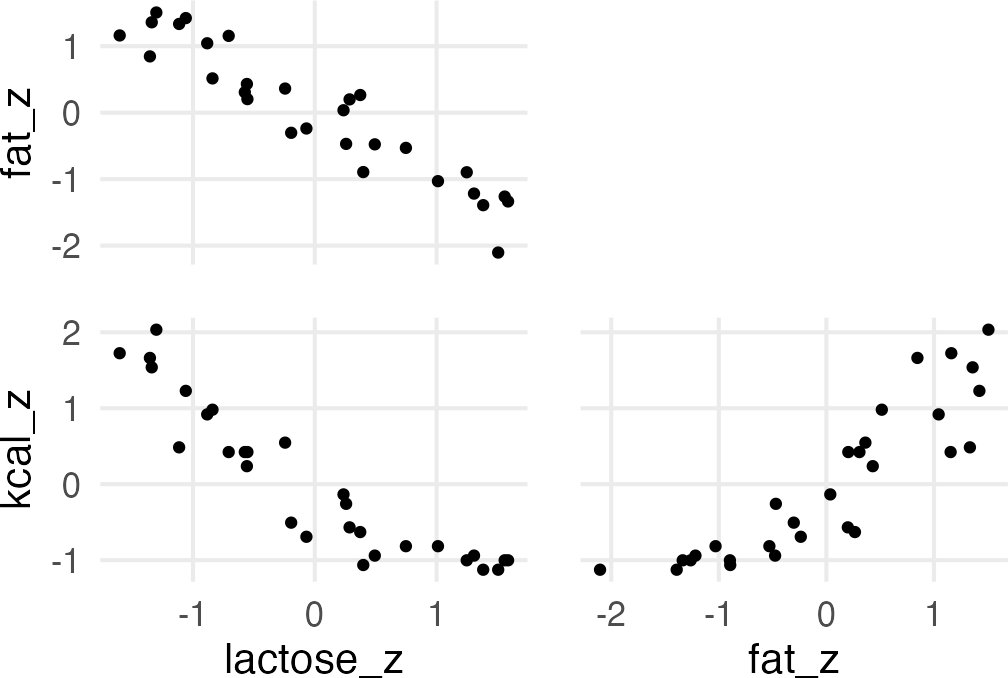
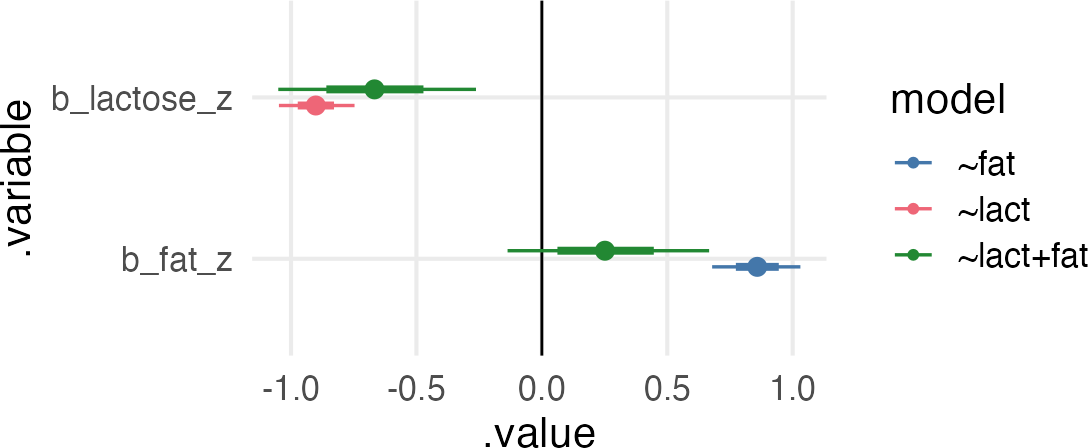
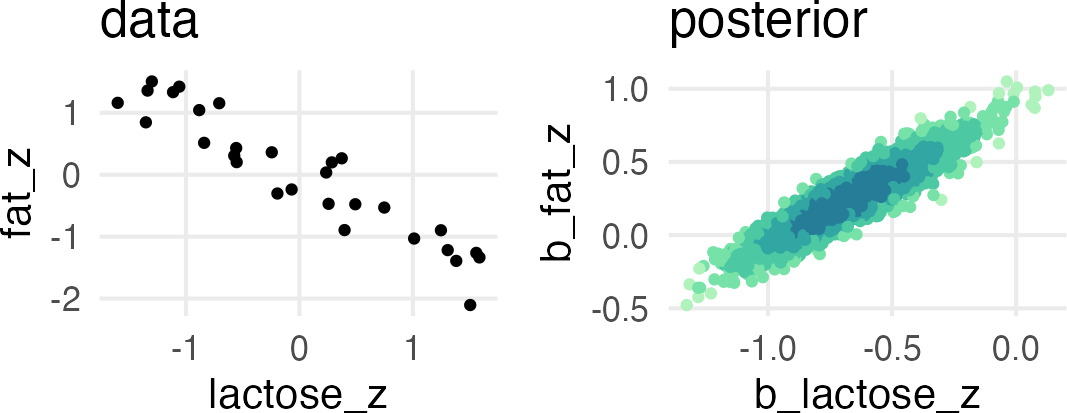
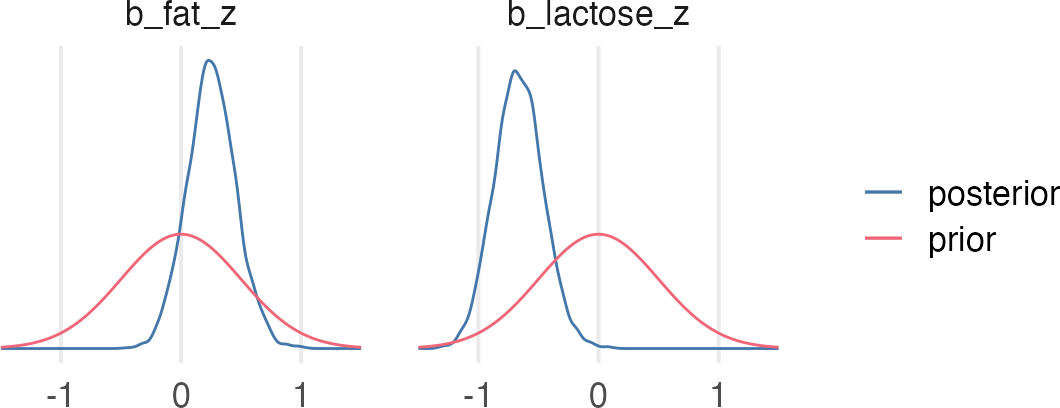
A real multicollinear example
A real multicollinear example involves the percent fat and lactose in primate’s milk when used to predict the kcal.
Making this work is going to involve both wrapping my mind around a post-treatment bias, and figuring out how to set a lognormal prior or family in brms.
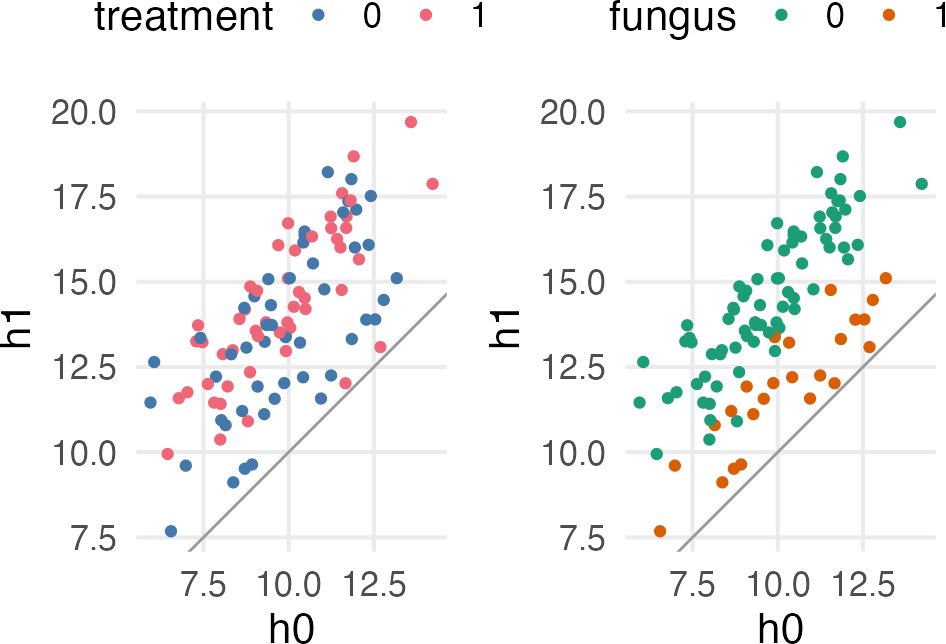
The hypothetical situation: You’re testing different antifungal soils on plant growth, and you’re measuring their height, and the presence/absence of fungus. The chronological process is something like:
flowchart LR
a[measure sprouts]
b(treat soil)
a --> b
c[measure plants]
d[record fungus]
b --> c
b --> d
The causal process might be something like
graph LR
h0[initial height]
h1[second height]
f[fungus]
t[treatment]
h0 --> h1
f --> h1
t --> f
This makes it much clearer now! “Post treatment” meaning “a variable that sits between the treatment and the outcome.”
fungus simulation
n =100tibble(plant_id =1:n,treatment = plant_id %%2,h0 =rnorm(n, 10, 2),fungus =rbinom(100,size =1,prob =0.5- treatment *0.4 ),h1 = h0 +rnorm( n, mean =5-3* fungus ))-> fungus_sim
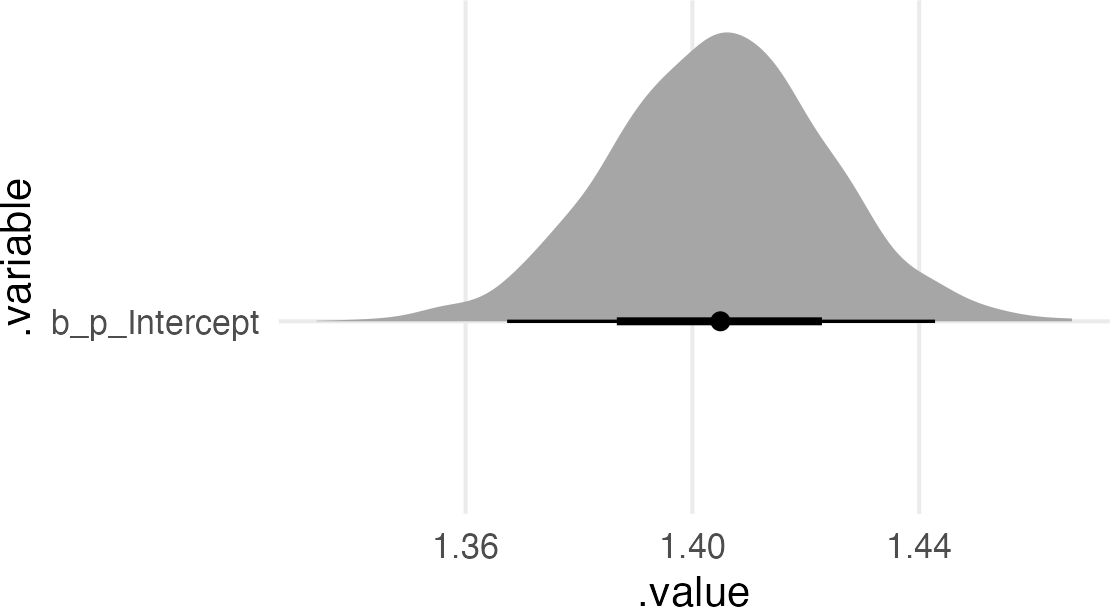
This is, thankfully, very similar to what the posterior from the book was! So maybe I did it right. Let’s grab the maximum likelihood estimate from the simulated data.
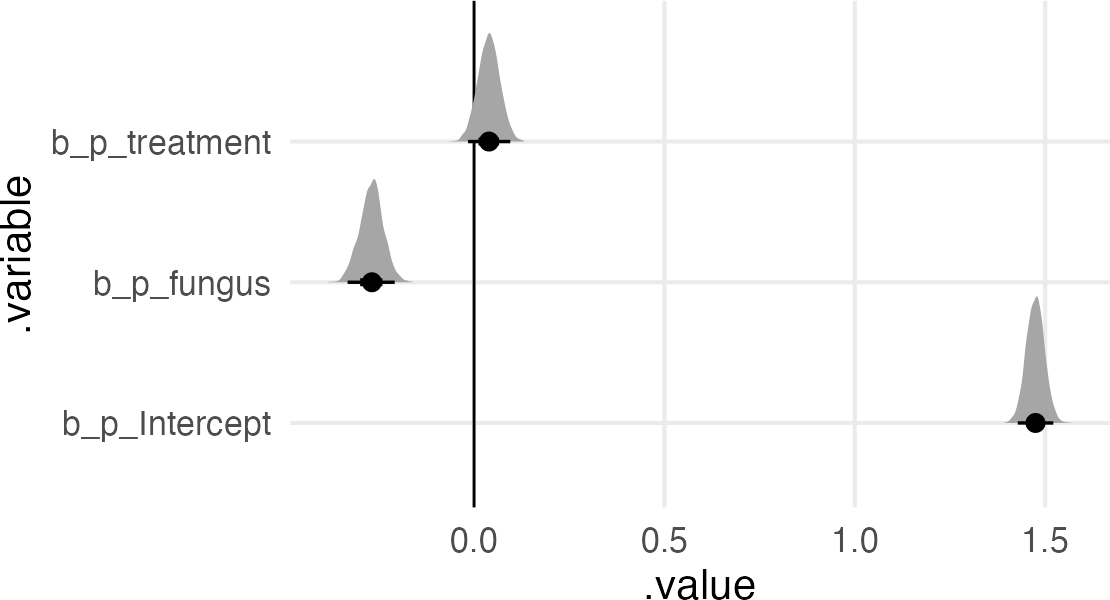
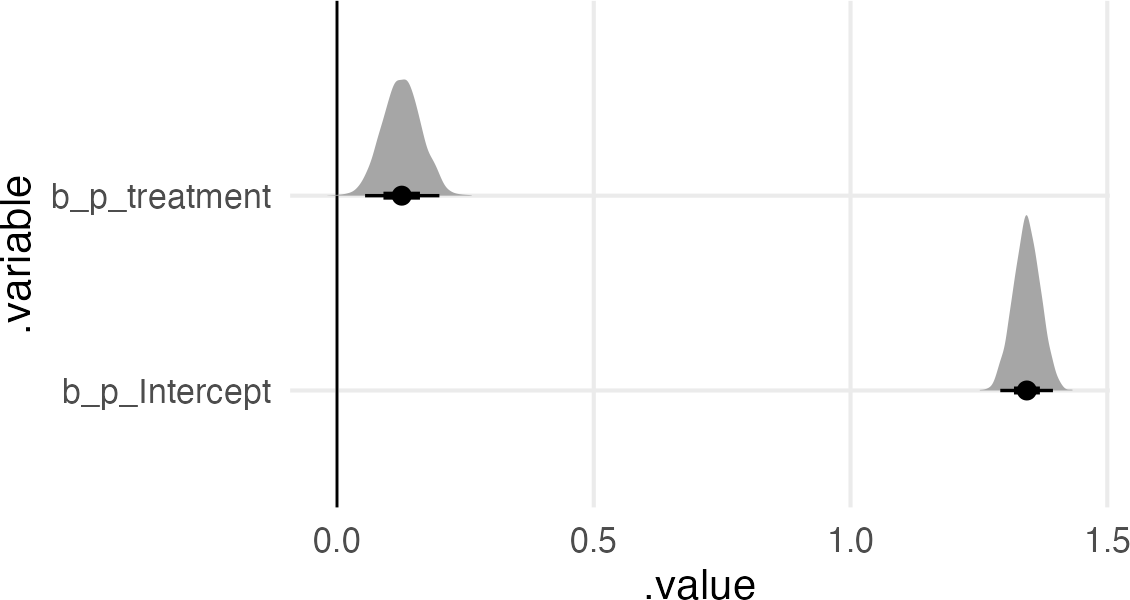
Now we’ll do the “bad” thing and include both predictors. The book keeps the lognormal prior on the intercept of the multiplier, but just a normal prior on the treatment and fungus effects.
The multiplier intercept got bigger (since it’s the growth for treatment=0, fungus=0).
We’ve got a negative effect of fungus.
We’ve got a weak or 0 effect of treatment.
The non-effect of treatment makes sense, since the effect of treatment is conditional on the effect of the fungus, and the presence/absence of fungus is itself an outcome of the treatment.
But, this doesn’t mean the treatment didn’t work. There are a lot more plants without fungus in the treatment condition than the non-treatment.
So, getting these conditional independence statements to look nice is a whole thing, apparently. There’s a unicode character, ⫫, but in LaTeX the best option is apparently \perp\!\!\!\perp, \(\perp\!\!\!\perp\).
This means that if fungus is included, then h1 (our outcome) is independent from treatment, i.e. including the post-treatment effect in the model will make it seem like there’s no effect of the treatment.